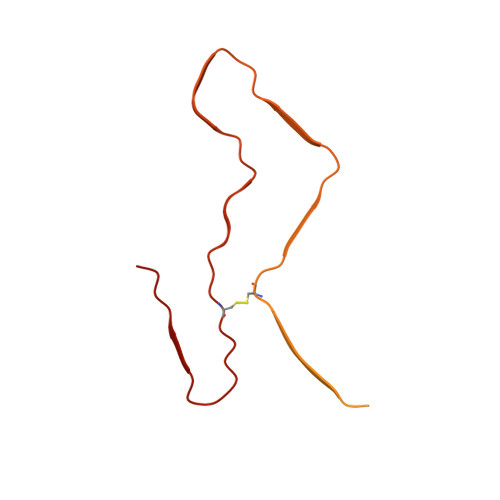
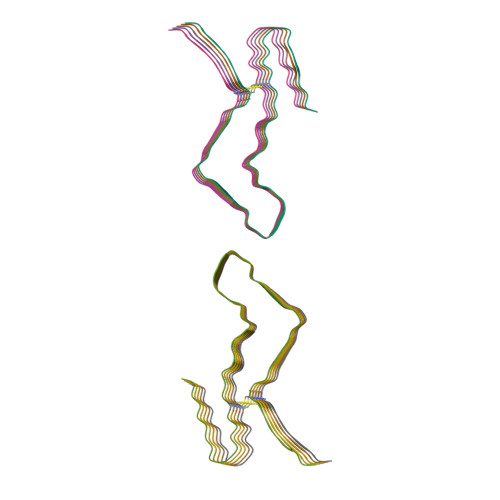Cryo-EM structure of an amyloid fibril formed by full-length human prion protein.
Wang, L.Q., Zhao, K., Yuan, H.Y., Wang, Q., Guan, Z., Tao, J., Li, X.N., Sun, Y., Yi, C.W., Chen, J., Li, D., Zhang, D., Yin, P., Liu, C., Liang, Y.(2020) Nat Struct Mol Biol 27: 598-602
- PubMed: 32514176
- DOI: https://doi.org/10.1038/s41594-020-0441-5
- Primary Citation of Related Structures:
6LNI - PubMed Abstract:
Prion diseases are caused by the misfolding of prion protein (PrP). Misfolded PrP forms protease-resistant aggregates in vivo (PrP Sc ) that are able to template the conversion of the native form of the protein (PrP C ), a property shared by in vitro-produced PrP fibrils. Here we produced amyloid fibrils in vitro from recombinant, full-length human PrP C (residues 23-231) and determined their structure using cryo-EM, building a model for the fibril core comprising residues 170-229. The PrP fibril consists of two protofibrils intertwined in a left-handed helix. Lys194 and Glu196 from opposing subunits form salt bridges, creating a hydrophilic cavity at the interface of the two protofibrils. By comparison with the structure of PrP C , we propose that two α-helices in the C-terminal domain of PrP C are converted into β-strands stabilized by a disulfide bond in the PrP fibril. Our data suggest that different PrP mutations may play distinct roles in modulating the conformational conversion.
Organizational Affiliation:
Hubei Key Laboratory of Cell Homeostasis, College of Life Sciences, Wuhan University, Wuhan, China.