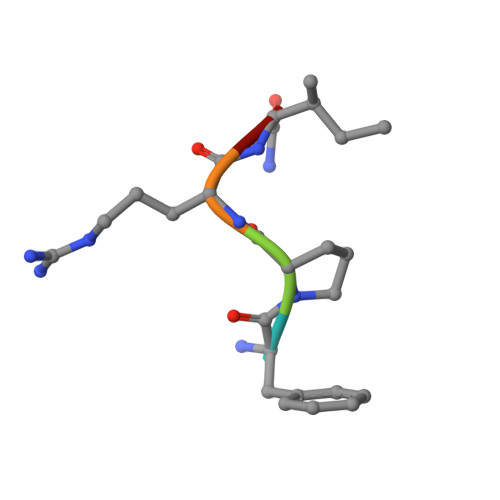
Rational design and characterization of D-Phe-Pro-D-Arg-derived direct thrombin inhibitors.
Figueiredo, A.C., Clement, C.C., Zakia, S., Gingold, J., Philipp, M., Pereira, P.J.(2012) PLoS One 7: e34354-e34354
- PubMed: 22457833 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1371/journal.pone.0034354
- Primary Citation Related Structures:
3U69, 3U8O, 3U8R, 3U8T - PubMed Abstract:
The tremendous social and economic impact of thrombotic disorders, together with the considerable risks associated to the currently available therapies, prompt for the development of more efficient and safer anticoagulants. Novel peptide-based thrombin inhibitors were identified using in silico structure-based design and further validated in vitro. The best candidate compounds contained both L- and D-amino acids, with the general sequence D-Phe(P3)-Pro(P2)-D-Arg(P1)-P1'-CONH₂. The P1' position was scanned with L- and D-isomers of natural or unnatural amino acids, covering the major chemical classes. The most potent non-covalent and proteolysis-resistant inhibitors contain small hydrophobic or polar amino acids (Gly, Ala, Ser, Cys, Thr) at the P1' position. The lead tetrapeptide, D-Phe-Pro-D-Arg-D-Thr-CONH₂, competitively inhibits α-thrombin's cleavage of the S2238 chromogenic substrate with a K(i) of 0.92 µM. In order to understand the molecular details of their inhibitory action, the three-dimensional structure of three peptides (with P1' L-isoleucine (fPrI), L-cysteine (fPrC) or D-threonine (fPrt)) in complex with human α-thrombin were determined by X-ray crystallography. All the inhibitors bind in a substrate-like orientation to the active site of the enzyme. The contacts established between the D-Arg residue in position P1 and thrombin are similar to those observed for the L-isomer in other substrates and inhibitors. However, fPrC and fPrt disrupt the active site His57-Ser195 hydrogen bond, while the combination of a P1 D-Arg and a bulkier P1' residue in fPrI induce an unfavorable geometry for the nucleophilic attack of the scissile bond by the catalytic serine. The experimental models explain the observed relative potency of the inhibitors, as well as their stability to proteolysis. Moreover, the newly identified direct thrombin inhibitors provide a novel pharmacophore platform for developing antithrombotic agents by exploring the conformational constrains imposed by the D-stereochemistry of the residues at positions P1 and P1'.
- IBMC-Instituto de Biologia Molecular e Celular, Universidade do Porto, Porto, Portugal.
Organizational Affiliation: