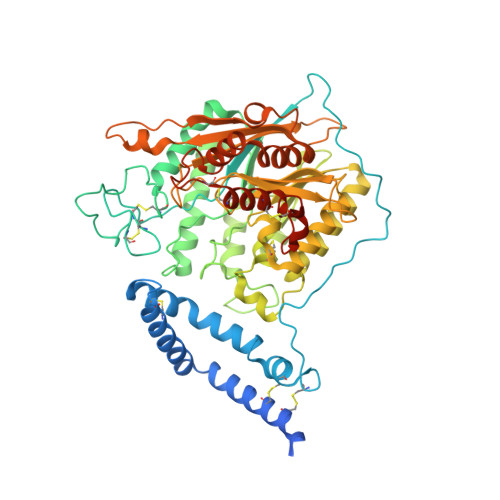
Human acid sphingomyelinase structures provide insight to molecular basis of Niemann-Pick disease.
Zhou, Y.F., Metcalf, M.C., Garman, S.C., Edmunds, T., Qiu, H., Wei, R.R.(2016) Nat Commun 7: 13082-13082
- PubMed: 27725636 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1038/ncomms13082
- Primary Citation Related Structures:
5I81, 5I85, 5I8R - PubMed Abstract:
Acid sphingomyelinase (ASM) hydrolyzes sphingomyelin to ceramide and phosphocholine, essential components of myelin in neurons. Genetic alterations in ASM lead to ASM deficiency (ASMD) and have been linked to Niemann-Pick disease types A and B. Olipudase alfa, a recombinant form of human ASM, is being developed as enzyme replacement therapy to treat the non-neurological manifestations of ASMD. Here we present the human ASM holoenzyme and product bound structures encompassing all of the functional domains. The catalytic domain has a metallophosphatase fold, and two zinc ions and one reaction product phosphocholine are identified in a histidine-rich active site. The structures reveal the underlying catalytic mechanism, in which two zinc ions activate a water molecule for nucleophilic attack of the phosphodiester bond. Docking of sphingomyelin provides a model that allows insight into the selectivity of the enzyme and how the ASM domains collaborate to complete hydrolysis. Mapping of known mutations provides a basic understanding on correlations between enzyme dysfunction and phenotypes observed in ASMD patients.
- Protein Engineering Department, Biologics Research, Sanofi, Framingham, Massachusetts 01701, USA.
Organizational Affiliation: