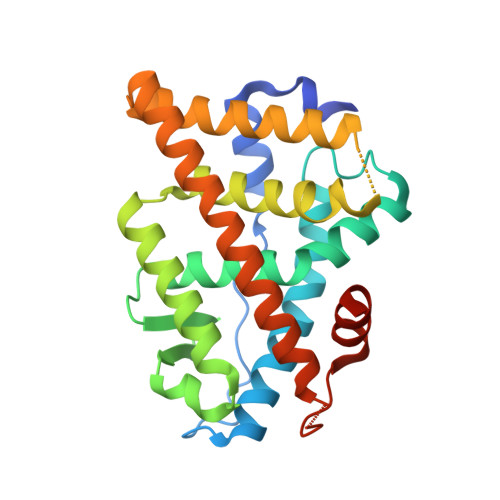
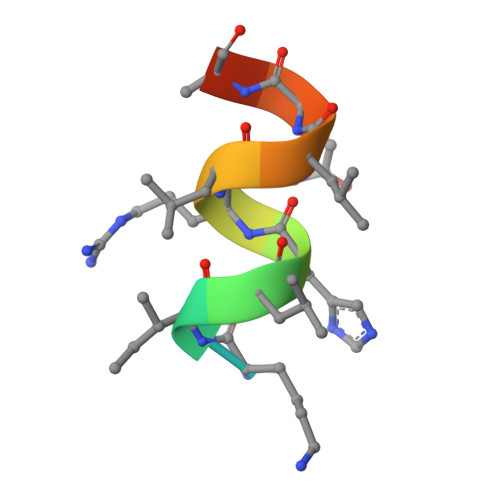
Design, synthesis, biological evaluation and crystal structure determination of dual modulators of carbonic anhydrases and estrogen receptors.
Tinivella, A., Nwachukwu, J.C., Angeli, A., Foschi, F., Benatti, A.L., Pinzi, L., Izard, T., Ferraroni, M., Erumbi, R., Christodoulou, M.S., Passarella, D., Supuran, C.T., Nettles, K.W., Rastelli, G.(2022) Eur J Med Chem 246: 115011-115011
- PubMed: 36516582 Search on PubMed
- DOI: https://doi.org/10.1016/j.ejmech.2022.115011
- Primary Citation Related Structures:
8EV1, 8EV2 - PubMed Abstract:
Multi-target compounds have become increasingly important for the development of safer and more effective drug candidates. In this work, we devised a combined ligand-based and structure-based multi-target repurposing strategy and applied it to a series of hexahydrocyclopenta[c]quinoline compounds synthesized previously. The in silico analyses identified human Carbonic Anhydrases (hCA) and Estrogen Receptors (ER) as top scoring candidates for dual modulation. hCA isoforms IX and XII, and ER subtypes ER⍺ and/or ERβ are co-expressed in various cancer cell types, including breast and prostate cancer cells. ER⍺ is the primary target of anti-estrogen therapy in breast cancer, and the hCA IX isoform is a therapeutic target in triple-negative breast cancer. ER⍺-mediated transcriptional programs and hCA activity in cancer cells promote favorable microenvironments for cell proliferation. Interestingly, several lines of evidence indicate that the combined modulation of these two targets may provide significant therapeutic benefits. Moving from these first results, two additional hexahydrocyclopenta[c]quinoline derivatives bearing a sulfonamide zinc binding group (hCA) and a phenolic hydroxyl (ER) pharmacophoric group placed at the appropriate locations were designed and synthesized. Interestingly, these compounds were able to directly modulate the activities of both hCA and ER targets. In cell-based assays, they inhibited proliferation of breast and prostate cancer cells with micromolar potency and cell type-selective efficacy. The compounds inhibited hCA activity with nanomolar potency and isoform-selectivity. In transactivation assays, they reduced estrogen-driven ER activity with micro-molar potency. Finally, crystal structures of the synthesized ligands in complex with the two targets revealed that the compounds bind directly to the hCA active site, as well as to the ER ligand-binding domain, providing structural explanation to the observed activity and a rationale for optimization of their dual activity. To the best of our knowledge, this work describes the design, synthesis and biological characterization of the first dual modulators of hCA and ER, laying the ground for the structure-based optimization of their multi-target activity.
- Department of Life Sciences, University of Modena and Reggio Emilia, Via Giuseppe Campi 103, 41125, Modena, Italy; Clinical and Experimental Medicine PhD Program, University of Modena and Reggio Emilia, Modena, Italy.
Organizational Affiliation: