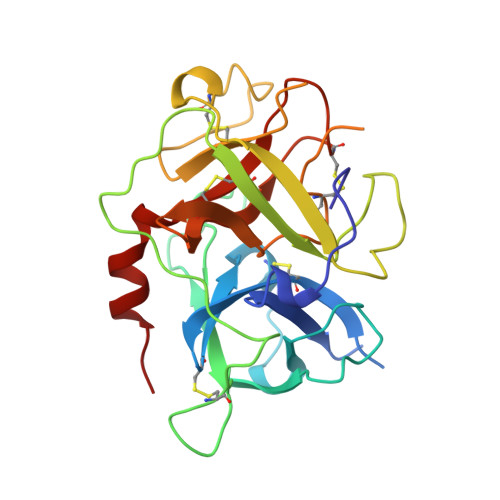
The 2.3 A crystal structure of the catalytic domain of recombinant two-chain human tissue-type plasminogen activator
Lamba, D., Bauer, M., Huber, R., Fischer, S., Rudolph, R., Kohnert, U., Bode, W.(1996) J Mol Biol 258: 117-135
- PubMed: 8613982 Search on PubMed
- DOI: https://doi.org/10.1006/jmbi.1996.0238
- Primary Citation Related Structures:
1RTF - PubMed Abstract:
Tissue-type plasminogen activator (t-PA), a multidomainal serine proteinase of the trypsin-family, catalyses the rate-limiting step in fibrinolysis, the activation of plasminogen to the fibrin-degrading proteinase plasmin. Trigonal crystals have been obtained of the recombinant catalytic domain of human-two-chain t-PA, consisting of a 17 residue A chain and the 252 residue B chain. Its X-ray crystal structure has been solved applying Patterson and isomorphous replacement methods, and has been crystallographically refined to an R-value of 0.184 at 2.3 A resolution. The chain fold, active-site geometry and Ile276-Asp477 salt bridge are similar to that observed for trypsin. A few surface-located insertion loops differ significantly, however. The disulfide bridge Cys315-Cys384, practically unique to the plasminogen activators, is incorporated without drastic conformational changes as the insertion loop preceding Cys384 makes a bulge on the molecular surface. The unique basic insertion loop Lys296-Arg304 flanking the primed subsites, which has been shown to be of importance for PAI-1 binding and for fibrin specificity, is partially disordered; it can therefore freely adapt to proteins docking to the active site. The S1 pocket of t-PA is almost identical to that of trypsin, whereas the S2 site is considerably reduced in size by the imposing Tyr368 side-chain, in agreement with the measured preference for P1 Arg and P2 Gly residues. The neighbouring S3-S4 hydrophobic groove is mainly hydrophobic in nature. The structure of the proteinase domain of two-chain t-PA suggests that the formation of a salt bridge between Lys429 and Asp477 may contribute to the unusually high catalytic activity of single-chain t-PA, thus stabilizing the catalytically active conformation without unmasking the Ile276 amino terminus. Modeling studies show that the covalently bound kringle 2 domain in full-length t-PA could interact with an extended hydrophobic groove in the catalytic domain; in such a docking geometry its "lysine binding site" and the "fibrin binding patch" of the catalytic domain are in close proximity.
- Max-Planck-Institut für Biochemie, Martinsried, Germany.
Organizational Affiliation: