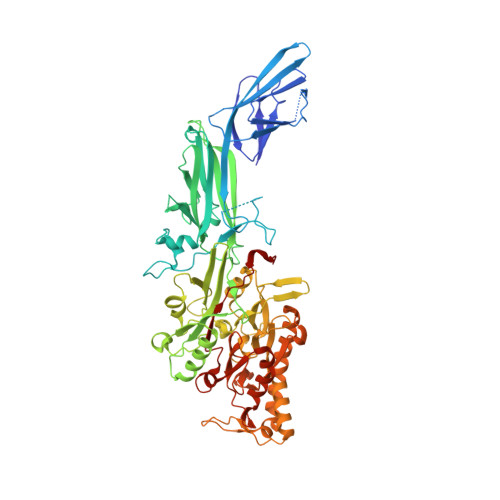
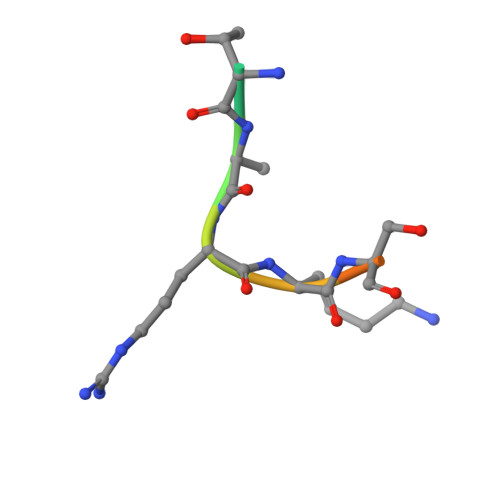
Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4
Arita, K., Shimizu, T., Hashimoto, H., Hidaka, Y., Yamada, M., Sato, M.(2006) Proc Natl Acad Sci U S A 103: 5291-5296
- PubMed: 16567635 Search on PubMedSearch on PubMed Central
- DOI: https://doi.org/10.1073/pnas.0509639103
- Primary Citation Related Structures:
2DEW, 2DEX, 2DEY - PubMed Abstract:
Histone arginine methylation is a posttranslational modification linked to the regulation of gene transcription. Unlike other posttranslational modifications, methylation has generally been regarded as stable, and enzymes that demethylate histone arginine residues have not been identified. However, it has recently been shown that human peptidylarginine deiminase 4 (PAD4), a Ca(2+)-dependent enzyme previously known to convert arginine residues to citrulline in histones, can also convert monomethylated arginine residues to citrulline both in vivo and in vitro. Citrullination of histone arginine residues by the enzyme antagonizes methylation by histone arginine methyltransferases and is thus a novel posttranslational modification that regulates the level of histone arginine methylation and gene activity. Here we present the crystal structures of a Ca(2+)-bound PAD4 mutant in complex with three histone N-terminal peptides, each consisting of 10 amino acid residues that include one target arginine residue for the enzyme (H3/Arg-8, H3/Arg-17, and H4/Arg-3). To each histone N-terminal peptide, the enzyme induces a beta-turn-like bent conformation composed of five successive residues at the molecular surface near the active site cleft. The remaining five residues are highly disordered. The enzyme recognizes each peptide through backbone atoms of the peptide with a possible consensus recognition motif. The sequence specificity of the peptide recognized by this enzyme is thought to be fairly broad. These observations provide structural insights into target protein recognition by histone modification enzymes and illustrate how PAD4 can target multiple arginine sites in the histone N-terminal tails.
- Field of Supramolecular Biology, International Graduate School of Arts and Sciences, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan.
Organizational Affiliation: